Requisitos y modelos de documentos
Requisitos para la solicitud de evaluación de estudios clínicos
Se recomienda, previamente a la presentación de la documentación completa, enviar un email a la dirección: ceic(ELIMINAR)@iislafe.es, adjuntando protocolo y copia de la aprobación de un CEIm acreditado en el territorio español, si se dispone.
Todos los estudios que vayan a realizar en el Hospital La Fe deberán contactar con administración_ec(ELIMINAR)@iislafe.es para la puesta en marcha y gestión de la documentación local.
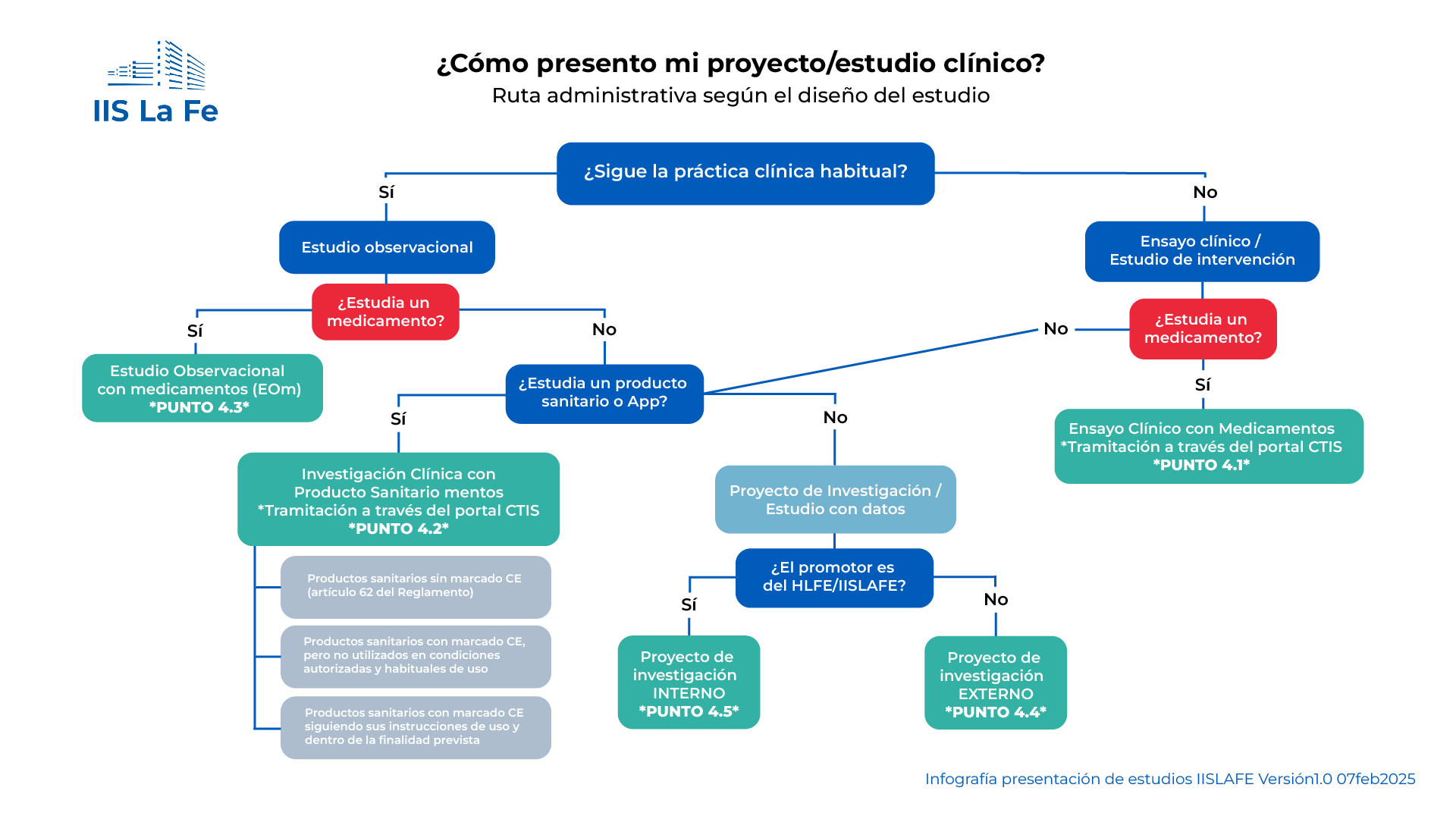
4.1. Evaluación de Ensayos Clínicos con Medicamentos
La documentación a presentar para la evaluación de ensayos clínicos con medicamentos/ productos sanitarios es la indicada en el "Documento de instrucciones de la Agencia Española de Medicamentos y Productos Sanitarios para la realización de ensayos clínicos en España".
Para los ensayos clínicos con medicamentos, el promotor debe presentar la documentación a través del Portal CTIS. Se deberá consultar antes al CEIm su disponibilidad para evaluarlo mediante un e-mail a la dirección: ceic@iislafe.es
4.2. Investigación clínica con Productos Sanitarios
El promotor debe presentar toda la documentación mediante un e-mail a la dirección ceic@iislafe.es. En dicho e-mail deben indicar el título del estudio, código de protocolo, promotor e Investigador Principal en el Hospital U. i P. La Fe.
Tipos de productos sanitarios:
En las investigaciones clínicas con productos sanitarios existen diferentes situaciones recogidas en la página de la AEMPS:
- Productos sanitarios sin marcado CE (artículo 62 del Reglamento).
- Productos sanitarios con marcado CE, pero no utilizados en condiciones autorizadas y habituales de uso.
- Productos sanitarios con marcado CE siguiendo sus instrucciones de uso y dentro de la finalidad prevista
Punto de contacto en la AEMPS para las investigaciones clínicas con productos sanitarios:
Tel.: +34 91 822 52 70. Correo electrónico: psinvclinic@aemps.es
DOCUMENTACIÓN ADICIONAL: Si el estudio cuenta con la aprobación de un CEIm acreditado en el territorio español, se adjuntará una copia del dictamen de aprobación.
4.2.1 Investigaciones clínicas con productos sanitarios sin marcado CE o con marcado CE, pero al margen del ámbito de su finalidad prevista. También se aplicaría a los estudios del funcionamiento con PSDIV que requirieran la autorización de la AEMPS (los cubiertos por el artículo 58 del reglamento 2017/746):
- Carta de presentación
- Protocolo completo. Podrá ser aceptado en inglés, con un resumen en la
lengua oficial del Estado. Se indicará su versión y fecha. - Cuaderno de recogida de datos.
- Aportación del certificado de marcado CE del producto sanitario.
- Ficha de indicaciones técnicas.
- Documento en relación con los Procedimientos y materiales utilizados para el
reclutamiento. - Hoja de información para los sujetos participantes y formulario de
consentimiento informado, o justificación de su exención. Se indicará su
versión y fecha. - Listado de investigadores de cada uno de los centros sanitarios en los que se
propone realizar el estudio y número de sujetos participantes que se pretenden
incluir en cada comunidad autónoma - Documentos a aportar de cada centro:
- Idoneidad de las instalaciones, según modelo publicado por la AEMP
- Compromiso y Curriculum vitae (CV) del investigador principal.
- Declaración de Conflicto de Intereses, si no consta en el CV.
- Formación en BPC.
- Fuentes de financiación del estudio y compensaciones previstas para los
sujetos participantes e investigadores, en su caso. - Certificado de la póliza de seguro en la que se indique la cobertura del estudi
o, así como los investigadores/centros asegurados, o garantía financiera para
la contratación de esta. - En caso de que la solicitud no la presente el promotor, esta deberá incluir un
documento que indique las tareas delegadas por el promotor a la persona o
empresa que actúa en su nombre. - Datos de facturación o solicitud de exención de tasas.
4.2.2 Investigaciones clínicas con productos sanitarios con marcado CE en las que el producto se va a utilizar dentro de su finalidad prevista y que SI requieren notificación a la AEMPS (artículo 74.1 del reglamento 2017/745 para PS y artículo 70.1 del reglamento 2017/746 para PSDIV):
- Carta de presentación
- Protocolo completo. Podrá ser aceptado en inglés, con un resumen en la lengua oficial del Estado. Se indicará su versión y fecha.
- Cuaderno de recogida de datos.
- Aportación del certificado de marcado CE del producto sanitario.
- Ficha de indicaciones técnicas.
- Documento en relación con los Procedimientos y materiales utilizados para el reclutamiento.
- Hoja de información para los sujetos participantes y formulario de consentimiento informado, o justificación de su exención. Se indicará su versión y fecha.
- Listado de investigadores de cada uno de los centros sanitarios en los que se propone realizar el estudio y número de sujetos participantes que se pretenden incluir en cada comunidad autónoma
- Documentos a aportar de cada centro:
- Idoneidad de las instalaciones, según modelo publicado por la AEMP
- Compromiso y Curriculum vitae (CV) del investigador principal.
- Declaración de Conflicto de Intereses, si no consta en el CV.
- Formación en BPC.
- Fuentes de financiación del estudio y compensaciones previstas para los sujetos participantes e investigadores, en su caso.
- Certificado de la póliza de seguro en la que se indique la cobertura del estudi o, así como los investigadores/centros asegurados, o garantía financiera para la contratación de esta.
- En caso de que la solicitud no la presente el promotor, esta deberá incluir un documento que indique las tareas delegadas por el promotor a la persona o empresa que actúa en su nombre.
- Datos de facturación o solicitud de exención de tasas.
4.2.3 Investigaciones clínicas con productos sanitarios con marcado CE siguiendo sus instrucciones de uso y dentro de la finalidad prevista:
- Carta de presentación
- Protocolo completo. Podrá ser aceptado en inglés, con un resumen en la lengua oficial del Estado. Se indicará su versión y fecha.
- Hoja de información para los sujetos participantes y formulario de consentimiento informado, o justificación de su exención. Se indicará su versión y fecha.
- Aportación del certificado de marcado CE del producto sanitario.
- Ficha de indicaciones técnicas.
- Listado de investigadores de cada uno de los centros sanitarios en los que se propone realizar el estudio y número de sujetos participantes que se pretenden incluir en cada comunidad autónoma.
- Fuentes de financiación del estudio y compensaciones previstas para los sujetos participantes e investigadores, en su caso.
- Formulario de recogida de datos.
- En caso de que la solicitud no la presente el promotor, esta deberá incluir un documento que indique las tareas delegadas por el promotor a la persona o empresa que actúa en su nombre.
- Para todos aquellos estudios donde exista un procedimiento invasivo y se aleatorice a los pacientes deberá presentarse una de las siguientes opciones:
- Certificado de la póliza de seguro en la que se indique la cobertura del estudio, así como los investigadores/centros asegurados, o garantía financiera para la contratación de esta.
- Solicitud en relación a los proyectos de investigación que conlleven algún procedimiento invasivo.
- ANEXO 1. Compromiso de responsabilidad Adicional (complementario a la solicitud) para los casos indicados en el formulario.
- Datos de facturación o solicitud de exención de tasas.
- Currículo vitae del Investigador/a Coordinador/a
- Formación en Buena Práctica Clínica (BPC) del Investigador/a Coordinador/a
4.3. Evaluación de Estudios Observacionales con medicamentos
El promotor debe presentar toda la documentación mediante un e-mail a la dirección ceic@iislafe.es. En dicho e-mail deben indicar el título del estudio, código de protocolo, promotor e Investigador Principal en el Hospital U. i P. La Fe.
Documentación a presentar:
- Carta de presentación.
- Protocolo completo, adaptado en la medida de lo posible a la estructura y contenido que se detalla en el anexo I del Real Decreto 957/2020, de 3 de noviembre, por el que se regulan los estudios observacionales con medicamentos de uso humano. Podrá ser aceptado en inglés, con un resumen en la lengua oficial del Estado. Se indicará su versión y fecha.
- Hoja de información para los sujetos participantes y formulario de consentimiento informado, o justificación de su exención. Se indicará su versión y fecha.
- Listado de investigadores de cada uno de los centros sanitarios en los que se propone realizar el estudio y número de sujetos participantes que se pretenden incluir en cada comunidad autónoma. Si el estudio se prevé realizar en otros países, listado de países.
- Fuentes de financiación del estudio y compensaciones previstas para los sujetos participantes e investigadores, en su caso. En caso de tratarse de una investigación clínica sin ánimo comercial, el promotor deberá presentar una declaración responsable firmada por el promotor y por el investigador coordinador de que el estudio cumple con todas las condiciones referidas en el párrafo e) del artículo 2.2 del Real Decreto 1090/2015, de 4 de diciembre.
- Formulario de recogida de datos.
- En caso de que la solicitud no la presente el promotor, esta deberá incluir un documento que indique las tareas delegadas por el promotor a la persona o empresa que actúa en su nombre.
- Datos de facturación o solicitud de exención de tasas.
- Currículo vitae del Investigador/a Coordinador/a
- Formación en Buena Práctica Clínica (BPC) del Investigador/a Coordinador/a
DOCUMENTACIÓN ADICIONAL: Si el estudio cuenta con la aprobación de un CEIm acreditado en el territorio español, se adjuntará una copia del dictamen de aprobación.
4.4. Proyectos de Investigación promovidos por empresas o personal externo (Proyectos regulados por Ley de Investigación Biomédica)
El promotor debe presentar toda la documentación mediante un e-mail a la dirección ceic@iislafe.es. En dicho e-mail deben indicar el título del estudio, código de protocolo, promotor e Investigador Principal en el Hospital U. i P. La Fe.
DOCUMENTACIÓN GENERAL:
- Carta de presentación
- Protocolo completo. Podrá ser aceptado en inglés, con un resumen en la lengua oficial del Estado. Se indicará su versión y fecha.
- Hoja de información para los sujetos participantes y formulario de consentimiento informado, o justificación de su exención. Se indicará su versión y fecha.
- Listado de investigadores de cada uno de los centros sanitarios en los que se propone realizar el estudio y número de sujetos participantes que se pretenden incluir en cada comunidad autónoma.
- Declaración de cumplimiento de la Ley de Protección de Datos (modelo específico - ver carpeta anexa)
- Fuentes de financiación del estudio y compensaciones previstas para los sujetos participantes e investigadores, en su caso. En caso de tratarse de una investigación clínica sin ánimo comercial, el promotor deberá presentar una declaración responsable firmada por el promotor y por el investigador coordinador de que el estudio cumple con todas las condiciones referidas en el párrafo e) del artículo 2.2 del Real Decreto 1090/2015, de 4 de diciembre.
- Formulario de recogida de datos.
- En caso de que la solicitud no la presente el promotor, esta deberá incluir un documento que indique las tareas delegadas por el promotor a la persona o empresa que actúa en su nombre.
- Para todos aquellos estudios donde exista un procedimiento invasivo y se aleatorice a los pacientes deberá presentarse una de las siguientes opciones:
- Certificado de la póliza de seguro en la que se indique la cobertura del estudio, así como los investigadores/centros asegurados, o garantía financiera para la contratación de esta.
- Solicitud en relación a los proyectos de investigación que conlleven algún procedimiento invasivo.
- ANEXO 1. Compromiso de responsabilidad Adicional (complementario a la solicitud) para los casos indicados en el formulario
- Datos de facturación o solicitud de exención de tasas
- Compromiso del Investigador Principal (modelo específico - ver carpeta anexa)
- Currículo vitae del Investigador Principal (fechado y firmado)
- Visto bueno jefe de servicio/ adjunta del área de enfermería, independientemente de que participe enfermería o no (modelo específico - ver carpeta anexa) (NO PROCEDE en estudios retrospectivos o estudios de investigación básica en los que no hay proceso asistencial).
SI CUENTAN CON LA APROBACIÓN DE OTRO CEIm:
Se realizará una revisión de aspectos locales, para la cual debe adjuntarse:
- Carta de presentación firmada por el promotor.
- Protocolo completo. Podrá ser aceptado en inglés, con un resumen en la lengua oficial del Estado. Se indicará su versión y fecha.
- Hoja de información para los sujetos participantes y formulario de consentimiento informado, o justificación de su exención. Se indicará su versión y fecha.
- Declaración de cumplimiento de la Ley de Protección de Datos (modelo específico - ver carpeta anexa).
- Información económica / memoria económica (si no va incluida en el protocolo).
- Datos de facturación o solicitud de exención de tasas.
- Compromiso del Investigador Principal (modelo específico - ver carpeta anexa)
- Currículo vitae del Investigador Principal (fechado y firmado)
- Visto bueno jefe de servicio/ adjunta del área de enfermería, independientemente de que participe enfermería o no (modelo específico - ver carpeta anexa) (NO PROCEDE en estudios retrospectivos o estudios de investigación básica en los que no hay proceso asistencial).
4.5. Proyectos de investigación propios (internos) (Solo para investigadores del Hospital la Fe/ Hospital de Manises/ Grupos acreditados o Unidades Mixtas del IIS La Fe) (No aplica para investigadores externos o empresas)
Si el proyecto NO es propio, sino que es un proyecto multicéntrico promovido por investigadores de otro centro, por favor, se deben seguir los requisitos especificados en el punto 4.4.
Los investigadores deben presentar toda la documentación mediante un e-mail a la dirección: area_cientifica@iislafe.es
Documentación a presentar:
- Solicitud de autorización de inicio (modelo específico - ver carpeta anexa)
- Memoria original del proyecto (modelo específico - ver carpeta anexa)
- Hoja de Información al Paciente y Consentimiento Informado; o justificación de su exención (modelo específico - ver carpeta anexa)
- Declaración de cumplimiento de la Ley de Protección de Datos (modelo específico - ver carpeta anexa)
- Para todos aquellos estudios donde exista un procedimiento invasivo y se aleatorice a los pacientes deberá presentarse una de las siguientes opciones:
- Certificado de la póliza de seguro en la que se indique la cobertura del estudio, así como los investigadores/centros asegurados, o garantía financiera para la contratación de esta.
- Solicitud en relación a los proyectos de investigación que conlleven algún procedimiento invasivo.
- Compromiso de responsabilidad Adicional (complementario a la solicitud) para los casos indicados en el formulario.
- Compromiso del Investigador Principal, en formaciones académicas no puede ser el estudiante, debe ser su tutor o director con vinculación laboral con el Hospital la Fe/ Hospital de Manises/ Grupos acreditados o Unidades Mixtas del IIS La Fe (modelo específico - ver carpeta anexa)
- Curriculum vitae del Investigador Principal fechado y firmado
- Visto Bueno jefe de servicio/ adjunta del área de enfermería, independientemente de que participe enfermería o no (modelo específico - ver carpeta anexa) (NO PROCEDE en estudios retrospectivos o estudios de investigación básica en los que no hay proceso asistencial)
- Formulario de colaboración de otros servicios implicados en el estudio clínico, si no participa ningún servicio debe indicarse en este documento (modelo específico - ver carpeta anexa)
- Aceptación de colaboración de otros servicios implicados en el estudio clínico, si aplica (modelo específico - ver carpeta anexa)
Respuesta a aclaraciones
Además de realizar los cambios solicitados en los documentos correspondientes, es necesario que respondan a cada uno de los puntos/ preguntas del documento de aclaraciones en un documento aparte, de lo contrario no se evaluarán las respuestas.
Por favor, envíen la nueva versión de los documentos que se modifican con control de cambios (cambios marcados) para su evaluación, así como una copia "limpia" para archivar.
Los documentos modificados deberán ir identificados (tanto en el propio documento como en el archivo electrónico) con un número de versión y fecha, que será distinto al inicial, si lo hubiera.
La respuesta y la documentación modificada, si procede, debe remitirse a la dirección de correo electrónico ceic@iislafe.es
Modificaciones sustanciales
El medio de presentación de las modificaciones sustanciales es el mismo que el de los estudios nuevos. Si se trata de ensayos clínicos con medicamentos, la documentación se presentará a través de CTIS. Sin embargo, cuando se trate de cualquier otro tipo de estudio, distinto de los ensayos clínicos con medicamentos, la documentación se presentará mediante un e-mail, a la dirección ceic@iislafe.es. En dicho e-mail deben indicar el título del estudio, código de protocolo, promotor e Investigador Principal en el Hospital U. i P. La Fe.
Documentación a presentar:
- Carta de presentación, en la cual se debe incluir:
- Identificación de la modificación sustancial mediante una fecha y un número o código (Ejemplo: Modificación sustancial nº1 de fecha 05/09/2025).
- Un breve resumen de los cambios y la justificación.
- Nueva versión de los documentos que se modifiquen, identificados con una nueva versión y una nueva fecha. Los documentos se deben presentar con los cambios resaltados (utilizando el control de cambios de Word)
- Nuevos documentos, cuando proceda
- Documentos que justifiquen los cambios, cuando proceda
- Datos de facturación (NO PROCEDE PARA PROYECTOS DE INVESTIGACIÓN INTERNOS)
Requisitos de protección de datos
Documentación a aportar: Si el estudio emplea Inteligencia Artificial (IA), o análisis masivo de datos (Big Data):
- DOCUMENTO con Información para el Registro de Actividad de Tratamiento (RAT) de acuerdo con art. 30 RGPD y art. 31 LOPDGDD
- Nombre y datos de contacto del responsable y, en su caso, del corresponsable, del representante del responsable, y del delegado de protección de datos;
- Los fines del tratamiento de datos;
- Descripción de las categorías de interesados y de datos personales;
- Si se comunicarán los datos a terceros, y en su caso, las categorías de destinatarios a quienes se comunicarán los datos;
- Si habrá transferencias de datos personales a un tercer país o una organización internacional (fuera de la unión Europea), y en su caso, la identificación de dicho tercer país u organización internacional y la documentación de garantías adecuadas, de acuerdo con el Capítulo V del RGPD,
- Los plazos previstos para la supresión de cada categoría de datos;
- Descripción general de las medidas técnicas y organizativas de seguridad aplicadas.
- DOCUMENTO con la evaluación de impacto en la protección de datos de acuerdo con art. 35 del RGPD. Antes de crear el registro, debe hacerse una EIPD que incluirá:
- Una descripción sistemática de las operaciones de tratamiento previstas y de los fines del tratamiento, inclusive, cuando proceda, el interés legítimo perseguido por el responsable del tratamiento;
- Una evaluación de la necesidad y la proporcionalidad de las operaciones de tratamiento con respecto a su finalidad;
- Una evaluación de los riesgos para los derechos y libertades de los interesados
- Las medidas previstas para afrontar los riesgos, incluidas garantías, medidas de seguridad y mecanismos que garanticen la protección de datos personales, y a demostrar la conformidad con el Reglamento, teniendo en cuenta los derechos e intereses legítimos de los interesados y de otras personas afectadas.
- DOCUMENTO de cumplimiento del Reglamento de Inteligencia Artificial.
- Mención en el protocolo al Plan de gestión de datos, que incluye los procesos aplicados en todo el ciclo de vida del dato, por ejemplo, recogida, anonimización, conservación, agrupación, tratamiento estadístico, consolidación, transmisión, destrucción.
- Mención en el protocolo al funcionamiento y la clasificación del producto de IA que se utiliza o desarrolla.
Solicitud en relación al seguro
Solicitud en relación a los proyectos de investigación que conlleven algún procedimiento invasivo (siempre que se aleatoricen a los participantes en el estudio y se siga la metodología de un ensayo clínico).